

During the last decade, aberrant DNA methylation has been identified as a hallmark of human cancer and several studies have highlighted the potential of DNA methylation as a clinically or diagnostically relevant biomarker. In comparison to their normal healthy counterparts, cancer cells generally display DNA hypermethylation at specific CpG islands, but the actual mechanism that drives this so-called CpG island methylator phenotype (CIMP) remains poorly understood.
To profile the DNA methylation landscape of human T-cell acute lymphoblastic leukemia (T-ALL), we analyzed 109 T-ALLs together with 10 stages of normal T cell development, which are considered the normal human counterparts of this disease, by 850 EPIC arrays. Here, we show that CpG islands are hypermethylated in all human T- ALLs compared to their normal counterparts. We designed a DNA methylation signature that can distinguish two types of T-ALL, with low or high levels of CpG island hypermethylation. This profile is dominated by CpGs in promoters of PRC2 target genes. T-ALLs with high levels of CpG island hypermethylation show low levels of H3K27me3 and vice versa, resulting in gene repression in both subtypes of T-ALL by different mechanisms.
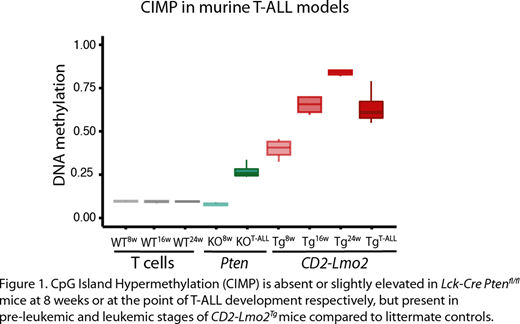
Furthermore, we found that aberrant CpG island hypermethylation shows a strong correlation with the epigenetic age of the leukemic T cells. By investigating the DNA methylation profile of two distinct mouse T-ALL models, the Lck-Cre Ptenfl/fland the CD2-Lmo2 transgenic mouse model, by Reduced Representation Bisulfite Sequencing, we could indeed recapitulate the DNA methylation features of the two human T-ALL subtypes in mice (Fig.1). The aggressive, fast-transforming Ptenfl/flmouse model displays low levels of CpG island hypermethylation, which correlated with human T-ALLs that have a shorter proliferative history and a worse prognosis. In contrast, murine CD2-Lmo2 T-ALLs have a longer disease latency and display a CpG island hypermethylation phenotype that is similar to human T-ALLs with a longer proliferative history. In CD2-Lmo2 mice, a pre-leukemic phase is present with self-renewing thymocytes. We elucidate that the CpG island methylation signature is gradually established in aging pre-leukemic thymocytes of 8, 16 and 24 weeks old CD2-Lmo2 mice (Fig.1). Of note, this hypermethylation phenotype is completely absent in age matched Lck-Cre Ptenfl/flmice that did not yet develop leukemia, suggesting that the proliferative history is responsible for aberrant CpG island DNA methylation observed in human T-ALL. Notably, this provides the first evidence that a pre-leukemic phase might be present in a large subset of human T-ALLs, and that epigenetic aberrations, either in the DNA methylation or histone methylation machinery are one of the first detectable alterations during T-ALL development.
Finally, using patient derived xenografts (PDX), we show that DNA hypomethylation by the FDA-approved hypomethylating agent Decitabine is very effective in treating T-ALL. Gene expression profiling revealed that the anti-leukemic effect is exerted by down-regulation of the oncogenic MYC pathway. However, by profiling these PDX T-ALLs by EPIC arrays, we unexpectedly uncover that the age-related CpG island hypermethylation signature is completely resistant to Decitabine treatment.
Altogether, our work demonstrates that DNA methylation reflects the epigenetic history of leukemic T cells and suggests that methylation-based subtypes of human T-ALL have followed a different trajectory towards T-cell transformation, possibly mediated by differences in the self-renewing capacity of the putative T-ALL cell-of-origin. Given that the concept of preleukemic thymocytes has only been reported in T-ALL mouse models so far, we here provide, for the first time, conceptual evidence that a pre-leukemic phase might also be involved in the pathogenesis of the human disease.
No relevant conflicts of interest to declare.

